

The ANDA process is how generic drug companies get legal permission in the U.S. to sell copies of brand-name medications. It’s not a shortcut-it’s a carefully designed legal and scientific pathway that saves patients billions while keeping safety strict. If you’re trying to bring a generic drug to market, understanding the legal requirements isn’t optional. It’s the foundation of everything.
What the ANDA Process Actually Is
The Abbreviated New Drug Application, or ANDA, was created by the Hatch-Waxman Act of 1984. This law was a deal: brand-name companies got extended patent protection, and generic companies got a faster, cheaper way to get their products approved. The word “abbreviated” doesn’t mean less rigorous-it means they don’t have to repeat the clinical trials the original drug already passed. The FDA already confirmed the brand drug is safe and effective. The generic just has to prove it’s the same.Today, 90% of prescriptions filled in the U.S. are for generics. That’s not luck. It’s the ANDA system working as intended. But getting there? It’s complex. Every step is governed by federal law, FDA regulations, and detailed guidance documents. Miss one requirement, and your application gets refused-no second chances without a full rewrite.
The Core Legal Requirements
To qualify for ANDA approval, your generic drug must meet five non-negotiable legal standards set by the FDA:- Identical active ingredient-No exceptions unless you file a suitability petition. Even a different salt form can trigger rejection.
- Same dosage form, strength, route, and conditions of use-If the brand is a 10mg tablet taken once daily, your version must be exactly that. No “improved” versions unless you go through the 505(b)(2) pathway.
- Bioequivalence-Your drug must perform the same in the body. This is proven through pharmacokinetic studies. The FDA requires the 90% confidence interval for Cmax and AUC to fall between 80% and 125% of the brand drug’s values. This isn’t a suggestion-it’s a legal threshold.
- Identical labeling-The prescribing information must match the brand, except for minor changes like the generic manufacturer’s name and the “AB” rating code. No adding warnings or removing contraindications.
- Comprehensive CMC data-Chemistry, Manufacturing, and Controls documentation must show you can consistently produce the drug to the same quality. This includes raw material specs, manufacturing processes, equipment validation, and stability data across multiple batches.
These aren’t guidelines. They’re legal requirements under Section 505(j) of the Federal Food, Drug, and Cosmetic Act. Failure to meet any one of them means your ANDA won’t be accepted-or worse, it gets approved and later recalled.
Submission Rules and Fees
The ANDA must be submitted electronically in eCTD format. Paper submissions are rejected outright. You also need to include Form FDA-356h (the application form) and Form FDA-3674 (the user fee cover sheet). There’s no way around this.As of 2024, the fee for a new ANDA is $129,500. That’s not optional. It’s a legal requirement under the Generic Drug User Fee Amendments (GDUFA). You pay it upfront, or your application doesn’t get reviewed. There’s also a $5,000 fee for any major changes after approval, called a prior approval supplement.
Manufacturing batches used in testing must be made at commercial scale-either 10% of the planned commercial batch size or 100,000 dosage units, whichever is larger. If your batch is too small, the FDA will refuse to review your application. This rule exists because small batches don’t prove you can make the drug consistently at scale.

Patent Certification and Legal Hurdles
One of the most complicated parts of the ANDA process is patent certification. You must choose one of four certifications:- Paragraph I-No patent listed for the drug.
- Paragraph II-The patent has expired.
- Paragraph III-You’ll wait until the patent expires.
- Paragraph IV-You believe the patent is invalid or won’t be infringed.
Paragraph IV is the most high-stakes. If you file it, the brand company has 45 days to sue you for patent infringement. If they do, the FDA can’t approve your drug for 30 months-or until the court rules, whichever comes first. This is called a “30-month stay.” Many generic companies delay filing Paragraph IV certifications because of this risk. In 2022, over 60% of ANDAs with Paragraph IV certifications faced litigation.
Brand companies have used patent thickets-filing dozens of minor patents on delivery systems, formulations, or methods of use-to block generics. The FDA’s 2023 Drug Competition Action Plan is trying to fix this, but legally, these tactics are still allowed. That’s why some generic approvals take five years, not three.
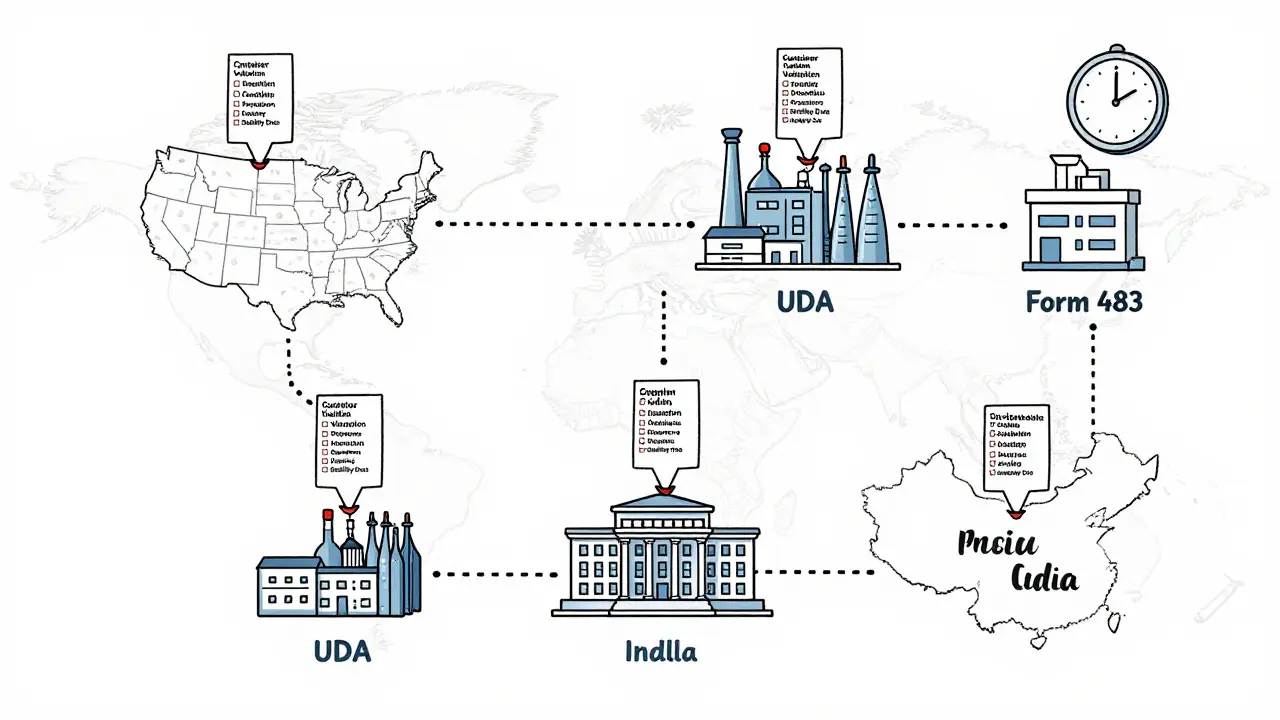
Manufacturing and Quality Control
Your manufacturing facility must follow Current Good Manufacturing Practices (cGMP). The FDA inspects every facility that produces an ANDA product-whether it’s in the U.S., India, or China. In 2022, 68% of FDA Form 483 observations (warning notices) were issued to overseas facilities.Common cGMP failures include:
- Inadequate container closure system validation
- Missing stability data for storage conditions
- Unvalidated cleaning procedures between batches
- Insufficient raw material testing
One regulatory affairs specialist on Reddit said their team had three ANDAs rejected because of poor container closure validation. That’s not rare. It’s standard. The FDA expects you to prove your packaging won’t degrade the drug over time. That means real-world testing under heat, humidity, and light-no shortcuts.
Why Some ANDAs Fail-And How to Avoid It
According to FDA data from 2022, 58% of first-time ANDA submissions get a deficiency letter. The top reasons:- 28%-Incomplete or flawed bioequivalence protocols
- 23%-Inadequate CMC documentation
- 15%-Missing or incorrect patent certifications
- 12%-Non-compliant eCTD format
Most failures happen because companies underestimate the depth of data needed. They think, “We copied the brand, so it should be fine.” But the FDA doesn’t care what you copied. They care what you proved.
Here’s how to avoid it:
- Use the FDA’s Orange Book to confirm the Reference Listed Drug (RLD) and its patent status.
- Run a pre-ANDA meeting with the FDA. In 2022, over 1,800 companies did this-and their approval rates jumped 30%.
- Hire a regulatory affairs professional with ANDA experience. The median salary for a certified RAC (Regulatory Affairs Certified) professional is $125,000. It’s worth it.
- Don’t rush CMC. Spend more time here than on bioequivalence studies. It’s the most common failure point.
How Long Does It Take?
The FDA’s goal under GDUFA III (2023-2027) is to approve 90% of standard ANDAs within 10 months. But reality is different. In 2022, the average review time was 36 months. Why? Complex generics-like inhalers, injectables, or topical creams-are taking longer. Only 42% of complex ANDAs get approved on the first try.Simple generics? They can move fast. Lupin got approval for a generic version of Jardiance in just 9.5 months because their application was clean and complete. Teva’s ANDA for a generic Advair Diskus took 42 months and cost $28 million extra because of device testing issues.
The timeline isn’t fixed. It depends on your product, your data, and how well you prepare.
What’s Next for the ANDA Process?
The FDA is investing $15 million in 2024 to develop better scientific tools for complex generics. They’re also testing AI to help review documents faster. The goal? Cut review times for simple ANDAs to under 24 months by 2027.But the biggest change may come from Congress. The Affordable Prescriptions for Patients Act of 2023 aims to limit patent thickets and make it harder for brand companies to block generics. If it passes, we could see a wave of new ANDAs approved faster.
For now, the ANDA process remains the most powerful tool we have to bring affordable drugs to patients. But it’s not easy. It’s legal, technical, and unforgiving. The companies that win aren’t the ones with the biggest budgets-they’re the ones who understand the rules inside and out.
Can a generic drug have a different inactive ingredient than the brand?
Yes, but only if the difference doesn’t affect safety or effectiveness. The FDA allows changes to inactive ingredients like colorants or fillers, as long as they don’t alter how the drug is absorbed or how it works. If a change could impact performance, the manufacturer must submit a prior approval supplement and prove it’s safe. Some companies use this to improve stability or reduce allergens, but they can’t change the active ingredient or dosage form.
What happens if my ANDA is rejected?
If the FDA refuses to receive your application, you get a letter listing exactly what’s missing. You can fix it and resubmit-but you’ll have to pay the user fee again. If your application is received but later rejected after review, you can respond to the deficiency letter with additional data. Most companies get at least one chance to fix issues. But if you ignore feedback or make the same mistakes twice, your application may be denied outright.
Do I need to own a manufacturing facility to file an ANDA?
No, you don’t need to own a facility, but you must have a contract manufacturer that’s FDA-inspected and compliant with cGMP. You’re still legally responsible for the quality of the product, even if you outsource production. The FDA will inspect the manufacturing site, not your office. Many small companies partner with contract manufacturers in India or the U.S., but they must document every step of the supply chain and maintain full control over quality.
Can I file an ANDA before the brand drug’s patent expires?
Yes, you can file an ANDA up to 4 years before the brand drug’s patent expires, as long as you submit a Paragraph IV certification. This allows you to get your application reviewed and approved in advance. But you can’t legally sell the drug until the patent expires or a court rules in your favor. This strategy is called “at-risk launch” and is common for high-demand drugs, but it carries legal risk if the patent is upheld.
Are all generic drugs approved through the ANDA process?
No. Some drugs use the 505(b)(2) pathway, especially if they’re modified versions-like a new delivery system or combination product. Biologics use a different pathway called 351(k). And over-the-counter (OTC) drugs follow OTC monographs, not ANDAs. Only traditional, chemically identical copies of brand-name prescription drugs go through the ANDA process. If your product changes how the drug works, you’re likely not eligible for an ANDA.












8 Comments